
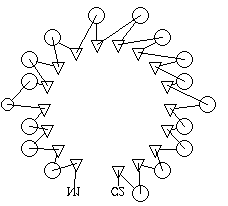
|
|
Constraints and bioinformatics
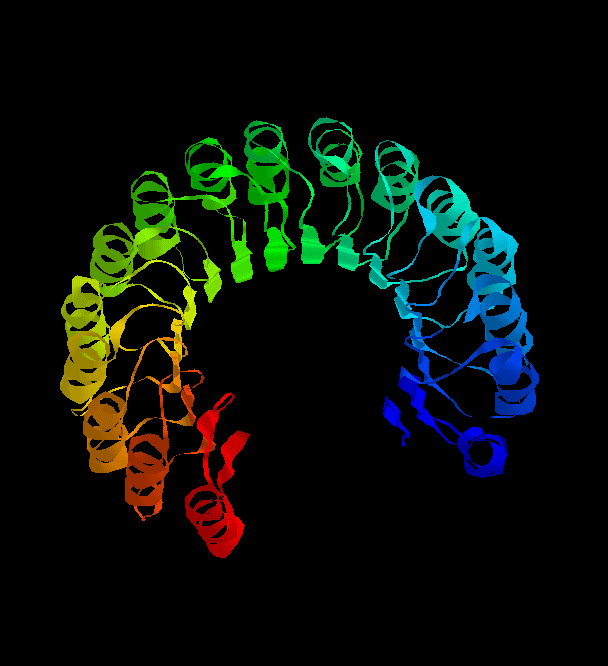
Protein Topology
Transcriptional regulatory regions
Biochemical networks and the analysis of gene expression data
Graph Database and Query Language for Bioinformatics
Interactive visualisation and exploration of biological data
Application of bioinformatics approaches to computer science
Neha Dhulia,
"ViewTOPS: A tool for visualising TOPS diagrams in 3D":
ViewTOPS will be a tool for visualising TOPS cartoons (topology representations of
protein structure). The project is based on work done by David Gilbert and David Weasthead on
"Formalising TOPS cartoons, the development of an interactive similarity search mechanism
over TOPS database and a TOPS-base structure comparison method".
Each TOPS cartoon represents Secondary Structure Elements (SSEs) and backbone of a
protein structure. It was originally represented as a pseudo-3D projection on a 2D
surface. This project aims to produce a system to display
the same cartoons in a custom-built 3D viewer.
This project is linked with the BBSRC/EPSRC funded TOPS project
"Patterns, functions and structures on a protein
topology database", joint between City University and the University of Leeds.
Darshit Mamtora,
"Database Of TOPS Patterns":
develop a TOPS "Motif" Database that will store patterns with biological
meaning and group them in a way that they are recognisable in the form of
sequences/families. These patterns will be determined from the result of a successful
match between the pattern(s) and structure(s) using the process of pattern
Discovery/Comparison.
This project is linked with the BBSRC/EPSRC funded TOPS project
"Patterns, functions and structures on a protein
topology database", joint between City University and the University of Leeds.
Tien Dung Vu,
"Visualisation, Pattern Discovery, and Pattern Matching of Topological Representation
of Transcriptional Regulatory Regions":
Design and implement a web-based system for Transcriptional Regulatory Regions
patterns. The system has a pattern database, graphic generation routines, pattern
discovery, pattern matching, web interface for humans, and web interface for programs.
This project is linked with
Dr Olivier Sand's European Union funded Marie Curie Fellowship project
"
Topological representation of transcriptional regulatory regions".
`the development and application of mathematical, statistical and computer methods
to analyse biological, biochemical and biophysical data, and guide biological
assays'.
This is very close to the NIH definition of Computational Biology:
Constraints and languages for description of biosequences:
We have formalised a language for describing constrained patterns in
biosequences, and developed an
efficient non-backtracking algorithm for finding a given pattern in a
sequence:
We have generalised this to a context-sensitive
language which permits the description of
two-dimensional
structure of biosequences, such as tandem repeats, stem
loops, palindromes and pseudo-knots.
Algorithms for structure searching using constraint logic programming and CSP
have been developed.
(In cooperation with Ingvar Eidhammer and
Inge Jonassen - University of Bergen, Norway),
[Sides: (Brazma
&
Gilbert) (Ratnayke's
implementation].
We have subsequently used constraint solving techniques in our
protein topology
pattern matching algorithm.
We have developed a constraint-based system for
searching protein databases, pattern discovery and protein structure
comparison. The approach is based on the TOPS topological
representation of protein structure, using the formal version of the
TOPS language we have made, and incorporates constraints over finite
domains. Searching is achieved using an efficient constraint-based
algorithm we have designed to match a TOPS pattern to a TOPS diagram.
We are now carrying out research into developing several approaches to
pattern
discovery in protein topology diagrams, and comparing
them.
The underlying motivation is to efficiently automatically generate
patterns classifying sets of proteins and to apply this to
characterising databases of protein structure.
We have developed a fast topology-based structure comparison method which is
not based on residues, but on secondary
structure elements (helices and strands) and associated H-bonds and chirality
information.
The system makes an alignment of the secondary structures of two proteins,
using a common pattern dicovered by our learning technique.
A score is calculated
which can be thought of as a measure of
the inserts-difference between the target and the diagram,
normalised by the size of the pattern discovered.
Computing issues:
These problems can be reduced to checking for subgraph isomorphism and finding
maximal common subgraphs in a restricted class of ordered graphs. We have
developed a subgraph isomorphism algorithm for vertex ordered graphs, which performs
well on the given set of data. The maximal common subgraph problem then is
solved by repeated subgraph extension and checking for isomorphisms. Despite
the apparent inefficiency such approach gives an algorithm with time
complexity proportional to the number of graphs in the input set and is still
practical on the given set of data.
In cooperation with David Westhead (University of Leeds), Janet Thornton
(EBI),
Nozomi Nagano (ex UCL)
and Juris Viksna (University of Latvia).
The project addresses the challenge of developing tractable methods
to analyse regulatory sequences in higher organisms, where strictly
sequence based approaches are less likely to succeed due to the complex
interaction of several regulatory elements. We propose to use a
topological representation of regulatory sites in order to take advantage
of the reduced level of complexity of algorithms that can be developed at
this level of abstraction. Topological approaches to the representation
and analysis of complex structures in bioinformatics have so far been
mostly developed in the field of protein structure. We propose to adapt
the work on TOPS diagrams carried out by David Gilbert to describe and
analyse transcriptional regulatory regions in genomes.
More information
here.
Computing issues:
As with TOPS,
checking for subgraph isomorphism and finding
maximal common subgraphs in a restricted class of vertex-ordered graphs.
An additional issue is that `vertices' have a length and these may overlap in
the order. We intend to use the approach developed in
[Brazma and Gilbert, 1995] and
[Eidhammer et al, 2001]
as a basis for the pattern language and (possibly) pattern matching algorithm.
Based on this data representation, we are developing prototype tools to
perform:
Computing issues:
Path searching algorithms in restricted types of graphs, checking for
subgraph isomorphism and finding
maximal common subgraphs in restricted classes of graphs. Automatic graph
layout.
See:
Computing issues:
expressivity, efficiency, data
manipulation and support for complex operations
such as pattern discovery.
Computing issues:
Analysis of higher order data. Automatic layout. Interactive displays.
Biological Issues:
The subgraph isomorphism and the maximal common
subgraph algorithms are used� for comparison and classification of
protein secondary structures, given as TOPS diagrams. They are
sufficiently fast to find in practical time a common substructure for any
given set of TOPS diagrams; moreover the checking of a given substructure
against the database of all TOPS diagrams can be done in miliseconds.
This approch can be used to speed up protein comparison at the secondary
structure level (the existing methods tend to be much slower) and to find
non-apparent common substructures. The limitation of this method is
comparatively small information content of part of the TOPS diagrams� -
good results can be obtained mainly for alpha-beta and beta proteins that
have sufficiently large number of secondary structure elements. The
current studies are focused on extension of TOPS formalism to made our
methods applicable to larger classes of prioteins. �
The project addresses the challenge of developing tractable methods
to analyse transcription regulatory regions in
higher organisms, where
strictly sequence
based approaches are less likely to succeed due to the complex interaction
of several regulatory elements. We propose to use a topological
representation
of regulatory sites in order to take advantage of the reduced level of
complexity of algorithms that can be developed at this level of abstraction.
Topological approaches to the representation and analysis of complex structures
in bioinformatics have so far been mostly developed in the field of
protein structure. We propose to adapt the work on
TOPS diagrams
carried out by
David Gilbert, to describe and
analyse transcriptional regulatory regions in genomes.
As with TOPS, checking for subgraph isomorphism and finding maximal common subgraphs in a restricted class of vertex-ordered graphs. An additional issue is that `vertices' have a
length and these may overlap in the order. We intend to use the approach developed in [Brazma and Gilbert, 1995] and [Eidhammer et al, 2001] as a basis for the pattern language and (possibly) pattern
matching algorithm.
Topological, abstract descriptions of proteins makes for faster structure
comparison. In turn, this allows for discovery of common patterns in sets
of structures. Rapid classification and possibly structure prediction are
the goals of this, the TOPS project.
Graph representations of some biological objects provide a richer
description than the usual sequence formats. We are preserving as much
three-dimensional information as possible, without slowing down the
subgraph isomorphism search algorithms. The discovery of patterns and
classification of new instances becomes feasible while avoiding the more
'brute force' methods of others.
Machine learning in protein topology: Beyond topological discovery.
Biological science is an "understanding" and "explanation" discipline. There
are few axioms or mathematical formulae to model biological systems.
Biological systems are highly complex and the only way to understand these
systems is by doing experiments. Thus, biological research is data driven
research. With the increase in data, biologists are unable to cope with the
expanding databases. Manual analysis and interpretation of the data is
impossible to carry out.
My research interest is in integrating the analysis from various biological
data (sequence, structure, and function) using several machine learning
techniques (including decision trees). Now, I'm looking at ways to combine
patterns discovered from different machine learning techniques.
A pattern is a set of common description express in high level language for a
class of objects. A motif is a pattern with biological meaning. E.g. Figure 1
for ferredoxin reductase protein family. Obviously, different motifs represent
different information for a group of proteins. Using combinations of "motifs"
, different levels of confidence can be assigned when characterising
a query protein into a protein family.
Figure 1: Ferredoxin reductase protein family motifs.
If you are a biologist you may understand the necessity for assigning
functions to genes but do not understand how to combine predictions with
varying levels of uncertainty. In contrast, if you are a computer
scientist, then you may understand how we can combine predictions with
varying levels of uncertainty but do not understand the biology of
assigning functions to genes.
Thus as a bioinformatician (the marriage of biology and computing) it is
my task to explain the reasoning behind this from a biologist point of
view and from a computer scientists point of view.
Biological Issues:
Thus due to the availability of the genome databases, we can use the
concept of homology to possibly annotate a few selective or random genes.
The sequences that we possibly may use in contributing to the homology
procedure are the following:
- Annotated sequences
A completed understanding and a more detailed view of this will be
available once the project reaches this stage.
People associated with the group:
What is Bioinformatics?
We regard Bioinformatics as an emergent discipline in its own right, and as a
new field has many definitions. We define Bioinformatics as
`the development and application of data-analytical and theoretical methods,
mathematical modeling and computational simulation techniques to the study of
biological, behavioral, and social systems.'
[
http://grants.nih.gov/grants/bistic/bistic.cfm]
Areas of our research:
Techniques that we use:
Research Projects
Past Research
Projects
Active Research Links:
David Gilbert's Research Interest
Constraints and bioinformatics
See
[Back-to-Top]
Protein Topology
[Back-to-Top]
Transcriptional regulatory regions
Olivier Sand (Marie Curie Research fellow), researcher.
[Back-to-Top]
Biochemical networks and the analysis of gene
expression data
In cooperation with
Jacques van Helden, Shoshana Wodak and Lorenz Wernisch, European Institute of
Bioinformatics.
EBI is currently developing a database of metabolic and regulatory
pathways, using an
entity-association model; the biochemical entities include proteins,
genes and small compounds. The associations represent processes by
which these entities interact with each other : reaction, catalysis,
activation/inhibition, expression, transcriptional regulation,
transport. This model amounts to formalising the cell as a large network
of interacting molecules. The network for yeast
includes some 6200 genes / proteins, and 4000 small compounds.
Such large networks
are split into several subgraphs, each encompassing a set of entities
that are involved in a common biological process (metabolic pathway,
signal transduction pathway, ...). Each subgraph is represented
graphically in the form of a pathway diagram.
[Back-to-Top]
Graph Database and Query Language for
Bioinformatics
The aim is to design the format of
a database suitable for bioinformatics data and to devise mechanisms to
perform
data deposit, retrieval and analysis. A prototype has been
developed to test
the implementation of database manipulation and query
mechanisms.
[Back-to-Top]
Interactive visualisation and exploration of biological
data
We are working on the display and navigation of biological data, using
distance metrics, clustering and various other algorithms (e.g.
spring-embedding).
This research is motivated by the
need to analyse the large and rapidly increasing amount of complex data
now available to researchers working in the field of bioinformatics.
We are presently undertaking the
design and development of an
experimental system.
Penny Noy, an industry funded research student, is undertaking research
into the visualisation of complex systems.
[Back-to-Top]
Application of bioinformatics approaches to
computer science
We have developed a theoretically founded framework for fuzzy unification and
resolution based on edit distance over trees. Our framework extends
classical unification and resolution conservatively. We prove
important properties of the framework and develop the FURY system,
which implements the framework efficiently using dynamic programming.
We evaluate the framework and system on a large problem in the
bioinformatics domain, that of detecting typographical errors in an
enzyme name database.
[Back-to-Top]
Juris Visksna's Research Interest
Automatic extraction of rules for protein classification
Computing Issues:� We have developed a new subgraph isomorphism
algorithm for vertex-ordered graphs. The algorithm is further used for
finding a maximal common subgraph by repeated extensions of subgraphs and
checking for isomorphisms with all graphs in the given set. This leads to
maximal common subgraph algorithm that works in linear time with respect
to the number of graphs in the given set; however the complexity grows
very rapidly with respect to the size of graphs - the algorithm appears
to be practical only for sparse graphs with up to 100 vertices and up to
100 edges. [Back-to-Top]
Olivier Sand's Research Project
Topological representation of transcription regulatory regions
In
order to function properly, our cells need to produce various definite
subsets of molecules in various precise amounts, depending on several
criteria. Those are location of the cell in the body (organ, tissue...),
age (foetus, infant, adult...), environment (temperature, infection,
drugs,...) and others. Some regions in our genome contain the information
to control the production of molecules by our cells. The understanding of
how these regulatory regions work
is essential for medical applications such as gene therapy.
Tools to identify and analyse the regulatory regions of genomes have been and
are being developped by bioinformaticians. My research project is a
participation to that effort.
[Back-to-Top]
Gilleain Torrance's Research
Project
Tops Project
[Back-to-Top]
Aik Choon Tan's Research
Project
BIOINFORMATICS: Machine Learning of Patterns for Protein Structures
and Functions.
[Back-to-Top]
Ali Al-Shahib's Research
Project
Assigning function to genes using a variety of data sources by
combining predication with varying levels of uncertainty
The research I'm proposing to do is assigning function to genes using a
variety of data sources and bioinformatics tools by combining predications
with varying levels of uncertainty.
There have been many debates regarding the exact number of
genes we have and whether or not 97% of our genome is 'junk'. Recently,
as a result of the completion of the human genome project and the
availability of the vast amount of databases, scientists have predicted
that we have 40,000 genes which means that we are literally 'twice a
worm'! There is also controvercies about how many control genes, how many
bacterial genes and how many mutations we have in our genome. This will
then help us in identifying whether or not we have 40,000 genes or whether
or not 97% of our genome is 'junk'. The main method into establishing
this is to functionally annotate the genes. By doing so, we can not only
establish the proportion of 'junk' we have in our genome but also
essentially contribute into the success of areas such as health,
agricultural, food and research industries.
- Genes having correlated patterns of expression
- Possible EST (Expressed Sequence Tags) or STS (Sequence Tag Sites)
related sequences
- Similar sequences in model organisms (e.g. E-coli or C.elegans)
As a result, we will finally obtain sequences that have varying levels of
Homology:
- Clear-function - Nucleotide/protein sequence has functional annotation
derived from probable homologues.
- Tentative-function - Nucleotide/protein sequence has functional
annotation derived from tentative homologues.
- Homologue - Nucleotide/protein sequence has probable homologues of
unknown function.
- No-homologue - Nucleotide/protein sequence has no probable homologues
Computing Issues:
After using the databases and the bioinformatics
tools to find possible homologues, we are proposing to program a computer
system to allow the user to access the annotated functions of the genes
easily and efficiently. As well as providing the user with a user-friendly
method of accessing the database of annotated functions, we are also
proposing to program the computer system to provide the user with an
understandable explanation for the particular annotation chosen. In order
to do so we must use the following techniques: 1- Weighted Logical Rules.
This is a more biologist friendly approach (Clark, 1993). After designing
a confidence level system whereby the sequence will be given an numerical
confidence level, logical rules will then be used to combine knowledge
from data sources which will limit the measures of uncertainty given by
each data source. 2- Bayesian Belief Network (BBN). A BBN is a graphical
network that represents probabilistic relationships among variables
(Haddawy, 1994). BBNs enable reasoning under uncertainty and combine the
advantages of an intuitive visual representation with a sound mathematical
basis in Bayesian probability (underlying theory) (Montironi, 1996). BBN
is not so much biologist friendly. Nevertheless, it has attracted much
recent attention as a possible solution for the problems of decision
support under uncertainty. [Back-to-Top]
Mallika Veeramalai's Research
Project
Incorporating sequence and interaction information in topological
models of protein structure
I am involved in algorithm development for TOPS Database to
enhance topological protein models with sequence and important
biochemical features (for eg. Ligand binding site, active site) that will
lead to structure-sequence-function relationships. Interesting results
would be valuable information to predict protein structure and function
from sequence, and these problems remain key challenges of direct
relevance to project in structural and functional genomics. [Back-to-Top]